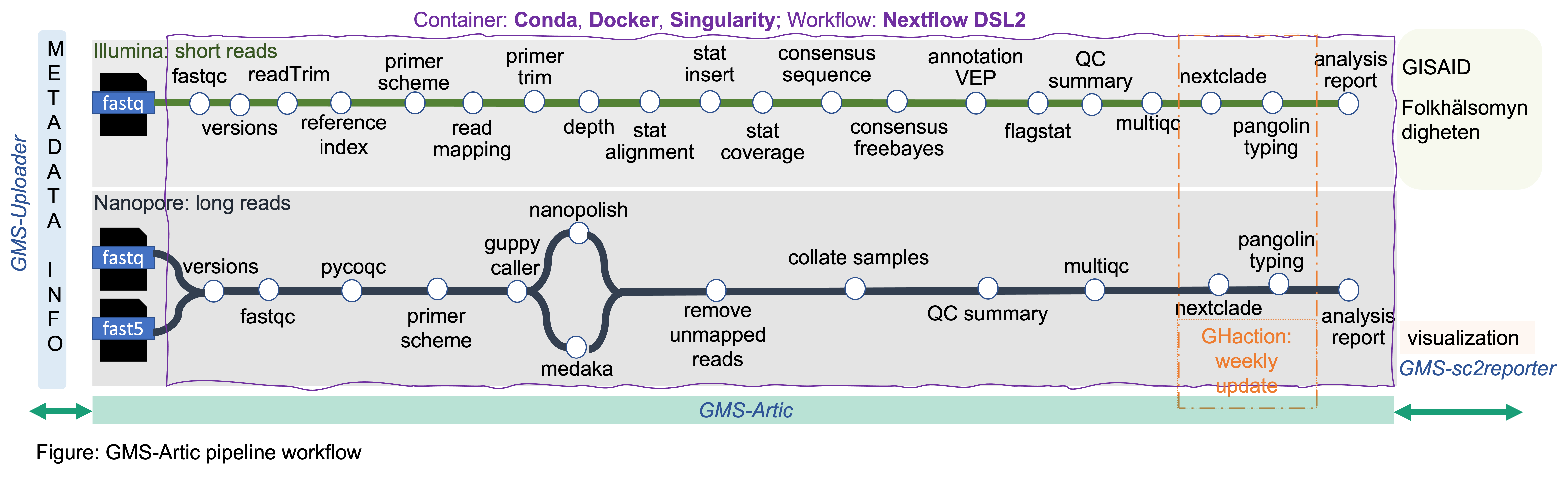
GMS-Artic
A nextflow pipeline with a GMS touch for running the ARTIC network’s fieldbioinformatics tools
What it does?
GitHub page

Major updates-v2.0.0
- Docker container separated for Pangolin typing
- Illumina container: gms-artic-illumina
- Nanopore container: gms-artic-nanopore
- Pangolin container: gms-artic-pangolin - bi-weekly updated with GitHub action
- Added separate package version files for each workflow
- versions: for Illumina and Nanopore
- pangoversion: for pangolin typing
- extra features for Illumina, flagstat, depth, VEP annotation
- Illumina results works for sc2reporter visualization
- additional QC features for Nanopore: fastqc, multiqc, pycoQC
How the results look?
example: Output of Illumina workflow run
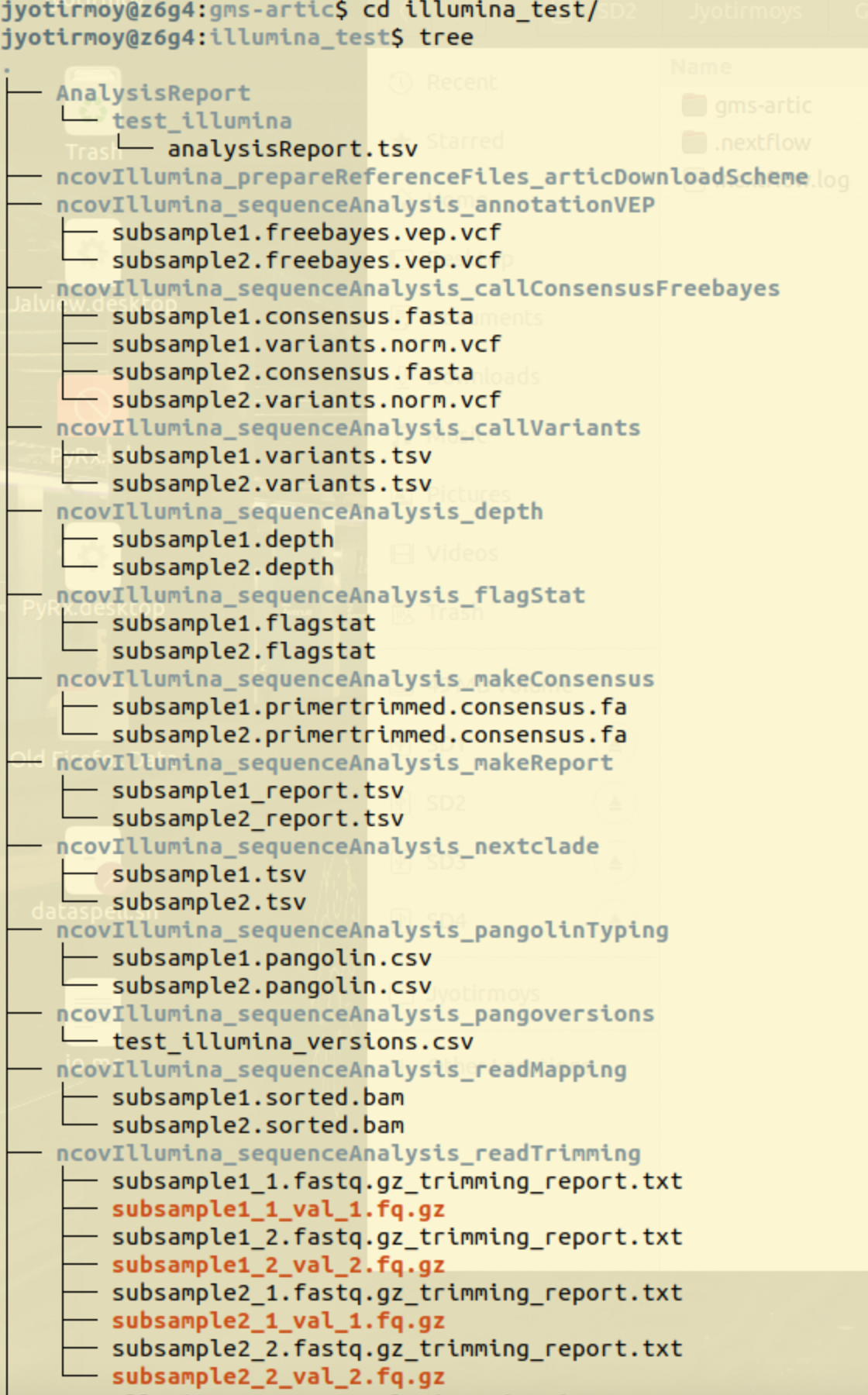
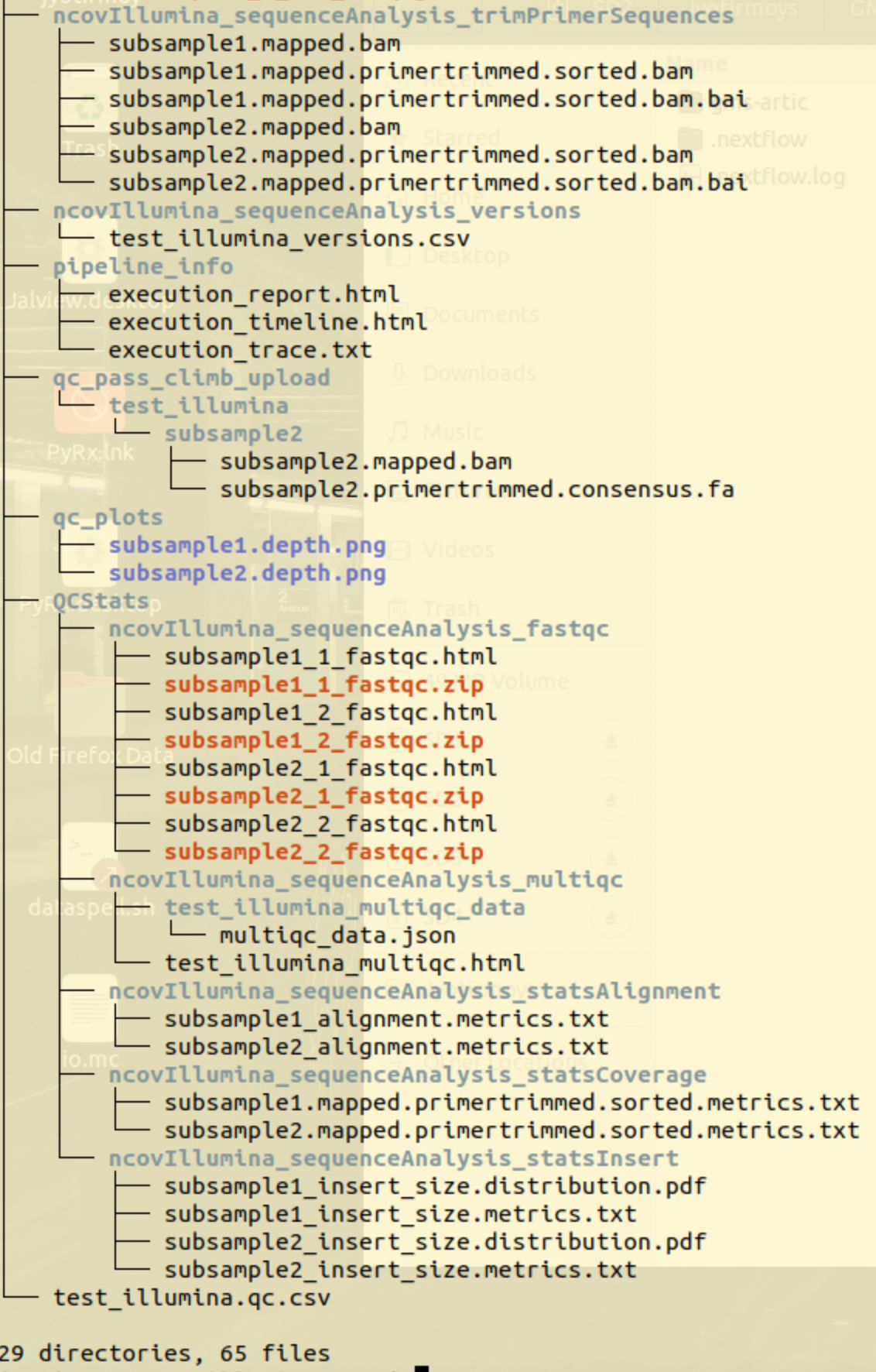
Where to run?
NGP-server @ Gothenburg University
- SGE cluster
- supports MPI

Local run?
local-server
- Singularity/Conda, Nextflow installation
- AMD64 OS arch (only AMD64 supported container)
Acknowledgements
- Genomic Medicine, Sweden
- Clinical Genomics:
- Gothenburg
- Linköping
- Lund
- Örebro
- Stockholm
- Umeå
- Uppsala
- all USERS of the pipeline!

![]()